21 CFR Part 4
Part 4 was added to 21 CFR, effective July 22, 2013. This is a new regulation, intended to help identify and clarify which rules apply to Combination Products. It provides regulatory framework and defines which Parts of 21 CFR apply to facilities that manufacture single-entity or co-packaged combination products.
In the past manufactures of Combination Products had to guess at what regulation(s) applied to their specific product. Often, manufactures opted to follow the Drug GMPs (Part 211), avoiding some of the messier portions of Part 820.
Part 4 clarifies matters by requiring that single-entity or co-packaged combination products must comply with the specifics of all relevant regulations. In most cases, this will mean that quality system for Combination Products will have to comply with both Parts 211 and 820.
Where requirements overlap, the FDA recommends following whichever guideline is more specific. The examples below help clarify the FDA’s recommendation:
|
Product |
Quality System |
cGPM Requirements |
QSR Requirements |
Recommended Guideline to Follow |
|
Drugs |
cGMP (Part 210 and 211) |
Specific requirements for calculation of yield (21 CFR 211.103) |
General requirement for calculation of yield as part of design validation (21 CFR 820.30) |
Drug cGMP (21 CFR 211.103) |
|
Device |
QSR (Part 820) |
General CAPA requirements identified as part of Production Record Review (21 CFR 211.192) |
Detailed CAPA requirements (21 CFR 820.100) |
QSR (21 CFR 820.100) |
However, when constituent parts are manufactured and marketed separately, only the respective requirement that applies to the constituent part must be followed, until the constituents are produced as a single-entity or co-packaged. For example, Company A produces a bulk drug product and Company B produces a delivery system for the bulk drug product. Company C co-packs the two products as a single product:
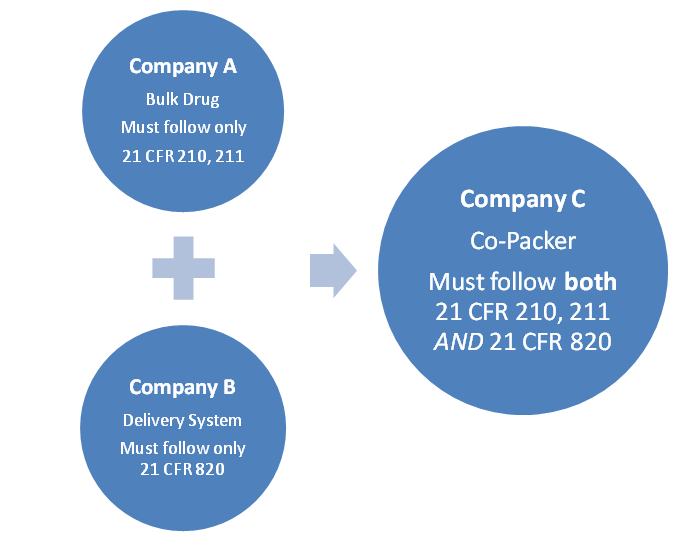
If an organization has already established a Quality System that is based on either cGMP or QSR requirements, there is a risk of being out of FDA compliance based on the new clarification provided in Part 4. Because maintaining 2 separate quality systems would be counterproductive, the FDA recommends expanding the existing systems to contain any missing elements. For example:
· When a combination drug and a device product is manufactured by a company with a Quality System designed on drug cGMPs, the requirements which are not duplicated in Part 820 must also be considered in the manufacturing process (e.g., CAPA, Design Controls, etc.)
-or-
· When a combination drug and device product is manufactured by a company with a Quality System designed on Part 820, the requirements which are not duplicated in Part 210 and 211 must also be considered in the manufacturing process, (e.g., Calculation of Yield, Tamper Evident Packaging, Stability Testing, etc.)
The clarity that Part 4 has provided may seem expensive and daunting from a Quality Systems perspective. However, revamping or completely overhauling an existing system is not necessarily required in light of the new requirements of Part 4. FDA compliance can be achieved by assessing an organization’s existing Quality Plan and simply filling in any gaps.